



The guidance is converging. The documentation expectations are clear. The question is whether your team is ready.
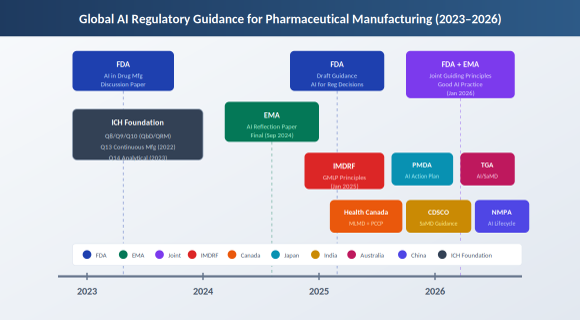
Figure 1. Global AI regulatory guidance timeline for pharmaceutical manufacturing (2023–2026)
The conversation about AI in pharmaceutical manufacturing has moved past the theoretical. Process engineers are running machine learning models against batch data. Quality teams are evaluating AI-assisted visual inspection systems. Data scientists are building predictive maintenance tools. The technology works. The harder question is whether the documentation and validation approach will hold up under regulatory scrutiny.
Between January 2025 and early 2026, the regulatory landscape shifted substantially. The FDA issued its first draft guidance on AI for regulatory decision-making. The EMA finalized its reflection paper on AI across the medicines lifecycle. The two agencies published joint guiding principles. Health Canada, PMDA, TGA, CDSCO, and NMPA each moved forward with their own frameworks. The result is a clearer picture than we had two years ago, though not a complete one.
This article maps the current state of AI guidance for CMC and manufacturing teams. It covers what the major regulators have said, where they align, and what the practical implications are for teams deploying AI in GMP environments.
FDA. In January 2025, the FDA published a draft guidance titled Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products [1]. This was the first FDA guidance to address AI use across the drug product lifecycle, including manufacturing. The document builds on CDER’s Framework for Regulatory Advanced Manufacturing Evaluation (FRAME) initiative and a March 2023 discussion paper specifically on AI in drug manufacturing [2].
The FDA’s approach centers on a credibility assessment framework. The agency asks three questions: What is the context of use? What is the risk if the model fails? And what evidence supports the model’s performance for that intended use? The framework is risk proportionate. A model used to flag potential deviations for human review carries different documentation expectations than a model that triggers automated rejection of product.
In practice: If you are using ML to identify golden batch parameters from historical data and the recommendations stay within your validated operating ranges, you document the model thoroughly but may not need a filing. If the ML output leads you to propose new parameter ranges outside your current registration, you are in SUPAC territory regardless of how you derived them.
The FDA has seen this before. CDER reports over 500 submissions with AI components since 2016, spanning nonclinical, clinical, post-marketing, and manufacturing phases [3]. The January 2025 guidance codifies what reviewers have been learning through those submissions.
EMA. The European Medicines Agency finalized its Reflection Paper on the Use of Artificial Intelligence in the Medicinal Product Lifecycle in September 2024, adopted by both the CHMP and CVMP [4]. The EMA takes a risk-based and human-centric approach. Regulatory impact is the trigger: AI uses that affect patient safety, efficacy assessment, or product quality require more rigorous documentation than uses that affect only the developer’s internal efficiency.
In practice: If you use AI to optimize your cleaning validation scheduling internally, the EMA considers this low regulatory impact. If you use AI to determine batch release decisions or to set specifications, the documentation requirements escalate. The EMA explicitly notes that AI tools must fit within existing quality system frameworks. This is not a separate compliance track.
FDA/EMA Joint Principles. In January 2026, the FDA and EMA jointly published ten guiding principles for Good AI Practice in Drug Development [5]. This was the first transatlantic regulatory alignment on AI in medicines. The principles cover human oversight, risk management, data governance, lifecycle controls, transparency, and accountability.
The joint principles are high-level by design. They establish a shared vocabulary and directional alignment without prescribing specific technical requirements. For CMC teams, the signal is clear: both agencies expect AI systems to be documented, validated, and monitored with the same rigor as any other GMP-critical system.
One notable element: the joint document acknowledges the problem of ‘shadow use,’ where analysts already work with large language models for daily tasks while leadership looks the other way. The principles push for better oversight and integration of data scientists with clinical and manufacturing leads throughout the product lifecycle.
Health Canada issued final Pre-Market Guidance for Machine-Learning Enabled Medical Devices in February 2025 [6]. The guidance includes a framework for Predetermined Change Control Plans (PCCPs), which allow manufacturers to pre-authorize defined model updates without requiring a new submission for each change. This concept is being adapted for drug manufacturing contexts as well.
In practice: A PCCP lets you define upfront that your predictive maintenance model will be retrained periodically (e.g., semi-annually or upon detection of performance drift) on new equipment data, within specified performance bounds, without filing each time. Without a PCCP framework, every model update could trigger a submission.
PMDA Japan published an Action Plan for AI in Operations in October 2025 [7], focused initially on the agency’s own use of AI in review and administrative tasks. Japan’s SaMD consultation desk provides early regulatory advice to developers. The country has also seen commercial deployment of federated AI models for drug discovery, reflecting a broader national push on AI in healthcare.
TGA Australia released updated guidance in February 2026 on when and how AI-based software as a medical device is regulated [8]. The TGA’s position is clear: regulatory scope is determined by the manufacturer’s intended purpose, not by the presence of AI features. If software uses AI to influence clinical decisions or patient care, it falls within the regulatory framework.
CDSCO India released a Draft Guidance Document on Medical Device Software in October 2025 [9]. This was the first Indian guidance to explicitly address AI/ML in medical applications. CDSCO requires transparent algorithm change-management protocols, documentation of model retraining, update triggers, and mechanisms to track performance drift. The guidance aligns India’s approach with IMDRF principles while addressing local registration requirements under the Medical Devices Rules, 2017.
In practice: For companies operating in India, the CDSCO draft guidance means AI/ML-based systems used in manufacturing quality decisions will need an Algorithm Change Protocol (ACP) documenting how model updates are validated before deployment. This is similar to the PCCP concept but with India-specific documentation requirements.
NMPA China published 2025 Measures for High-End Medical Device Lifecycle Regulation [10], which include AI medical devices as a priority area. The measures establish a conditional approval pathway for AI innovations pioneered in China and push for accelerated standards development. NMPA is also working to establish an AI medical device standardization organization.
IMDRF released final Good Machine Learning Practice principles in January 2025 [11], building on the 2021 guiding principles from FDA, Health Canada, and MHRA. These ten principles provide a harmonized global baseline for AI/ML-enabled medical devices and are increasingly referenced in drug manufacturing contexts.

The guidance documents address AI across the product lifecycle, but for CMC and manufacturing teams, the most relevant applications fall into several categories. Each has different regulatory implications depending on how the AI output is used.
Process optimization and golden batch analytics. Machine learning models analyze historical batch data to identify parameter combinations that correlate with higher yield, better quality, or improved productivity. The output is typically prescriptive recommendations for process control strategy adjustments.
Application example: Analyzing 40 historical batches to identify which parameter trajectories correlate with top-quartile yield, then tightening control limits within existing validated ranges. This stays within process knowledge if no registered parameters change.
Real-time release testing and PAT integration. AI/ML models support real-time decisions about product quality based on process analytical technology data, potentially reducing or eliminating end-product testing for specific attributes.
Application example: Using NIR spectroscopy with an ML model to predict API content in tablets during compression, eliminating the need for HPLC release testing on that attribute. This requires regulatory approval because it changes the registered release method.
Predictive maintenance and deviation forecasting. Models trained on equipment performance data predict failures before they occur or flag anomalies that may indicate emerging deviations.
Application example: Monitoring bioreactor agitator vibration signatures to predict bearing failure weeks in advance, allowing scheduled replacement during planned downtime. This is typically internal operational improvement with minimal regulatory impact.
Visual inspection and defect detection. Automated visual inspection systems using machine learning classify defects in filled containers, tablets, or other product forms.
Application example: Replacing manual inspection of vials with an ML-based system that detects particulates, cracks, and fill-level deviations. This requires validation as a process change and typically a variation filing.
The common thread across all these applications: they require documentation of the model’s intended use, training data, validation approach, performance monitoring, and change control. The technology may be novel, but the quality system expectations are familiar.
The question CMC teams ask most often is not whether AI can be used, but whether using it will trigger a regulatory filing. The answer depends on what the AI output does.
The key distinction is between process knowledge and regulatory decision-making. If the ML output informs your internal understanding of the process but does not change registered parameters, specifications, or control strategies, it falls into the process knowledge category. You document it thoroughly, you use it to improve operations, but you do not necessarily file a supplement or variation.
If the ML output triggers a change to your control strategy, your filed process parameters, or your release specifications, you are in change control territory. The change itself determines the filing pathway, not the fact that ML was involved. A parameter range change justified by ML analysis follows the same SUPAC or variation logic as a parameter range change justified by any other process development work.
Practical decision framework:
1. Does the AI output change any registered process parameter, specification, or control strategy? If no, document as process knowledge under continuous process verification.
2. If yes, does the change fall within an existing approved design space or flexibility provision? If yes, implement under internal change control. Note that intra-design-space changes are generally managed without prior regulatory approval, though regional requirements vary: the EMA treats these as non-notifiable, while the FDA may require documentation in annual reports depending on how established conditions were defined per ICH Q12.
3. If the change exceeds registered flexibility, follow your standard post-approval change pathway (SUPAC, variation, amendment) based on the nature of the change, not the nature of the tool that identified it.
Both the FDA’s Emerging Technology Program and the EMA’s Innovation Task Force offer pathways for early engagement [12]. If you are deploying AI in a novel manufacturing context and are unsure how it will be received, these programs allow informal dialogue with reviewers before you commit to a filing strategy.
The regulatory framework applies equally to innovator and generic manufacturers, but the commercial calculus is different.
Innovator companies typically engage AI/ML during process development, using models to define design space, optimize scale-up, and support continuous process verification. The regulatory conversation happens early, often as part of the original submission or through early engagement programs.
Generic manufacturers face a different situation. Before reaching the manufacturing stage, generic drug developers must conduct bioequivalence studies — abbreviated clinical trials demonstrating that the generic product matches the reference drug’s rate and extent of absorption. Biosimilars require more extensive clinical development, including analytical similarity assessments, functional assays, PK/PD studies, and clinical immunogenicity studies. AI/ML tools are increasingly used during this development phase for formulation optimization and in silico bioequivalence modeling.
Once approved, the process is validated and the ANDA is filed. The question then becomes whether retrospective analysis of manufacturing data can yield productivity improvements without triggering a post-approval filing or compromising the validated state. This is where golden batch analytics and similar approaches offer particular value.
The zero-capital value proposition matters for generics. Margin pressure is real. If machine learning can identify that tighter control of a specific parameter correlates with higher yield, and that tighter control is achievable within existing validated ranges using existing equipment, the productivity gain costs nothing but the analysis work. This is process understanding, not process change, and it can be documented accordingly.
The regulatory framework does not distinguish between innovator and generic for AI documentation purposes. But the business case, and the practical approach, often looks different.
The regulatory posture that works is the same one that has always worked: build process understanding, document rigorously, file when the data demands it. AI/ML does not change that equation. It changes the tools available for building understanding and the volume of data you can analyze, but not the fundamental expectation that you know your process and can defend your decisions.
The guidance is converging globally. The FDA/EMA joint principles signal that transatlantic alignment is a priority. Health Canada, PMDA, TGA, CDSCO, and NMPA are each moving in broadly compatible directions. The principles-based approach means detailed technical requirements will continue to develop, but the direction is clear.
For teams deploying AI in manufacturing, the practical steps are straightforward. Define the intended use. Document the training data and model development. Validate performance for the specific context. Monitor ongoing performance. Apply change control when the model or its use changes. Engage regulators early if the application is novel or high-risk.
The next article in this series will walk through a case study: golden batch analytics in biologics manufacturing. It covers the two-stage ML approach, unsupervised clustering followed by supervised profiling, the prescriptive recommendations that emerged, and how the work was framed to stay within process knowledge rather than triggering a filing.
[1] FDA. Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products. Draft Guidance. January 2025.
[2] FDA/CDER. Artificial Intelligence in Drug Manufacturing. Discussion Paper. March 2023.
[3] FDA. Artificial Intelligence for Drug Development. CDER Website. Updated February 2025.
[4] EMA. Reflection Paper on the Use of Artificial Intelligence (AI) in the Medicinal Product Lifecycle. EMA/CHMP/CVMP/83833/2023. September 2024.
[5] FDA/EMA. Guiding Principles of Good AI Practice in Drug Development. Joint Publication. January 2026.
[6] Health Canada. Pre-Market Guidance for Machine-Learning Enabled Medical Devices. February 2025.
[7] PMDA. Action Plan for the Use of AI in Operations at the PMDA. October 2025.
[8] TGA. Artificial Intelligence (AI) and Medical Device Software Regulation. Guidance. February 2026.
[9] CDSCO. Draft Guidance Document on Medical Device Software. October 2025.
[10] NMPA. Measures for High-End Medical Device Lifecycle Regulation. Announcement No. 63. July 2025.
[11] IMDRF. Good Machine Learning Practice for Medical Device Development: Guiding Principles. Final Document. January 2025.
[12] FDA. Emerging Technology Program. CDER Website; EMA. Innovation Task Force. EMA Website.
Written from field experience supporting AI/ML deployment in pharmaceutical manufacturing and regulatory review. Regulatory citations are current as of June 2026. Confirm against the latest guidance documents for your specific context.

Passionate biopharma professional and founder of Bio Pharma View, dedicated to sharing industry knowledge, practical experiences, and insights from the pharmaceutical and biotechnology sector.


